
Research areas
- aPhysiological patterns of activity in the developing brain
- bNetworks organization in the developping primary somatosensory cortex
- cDepolarizing actions of GABA in development and disease
- dDevelopmental brain plasticity
- eHealthy and pathological brain & exercise
- fPost traumatic depression and reactive plasticity
Physiological patterns of activity in the developing brain
RUSTEM KHAZIPOV & MARAT MINLEBAEV
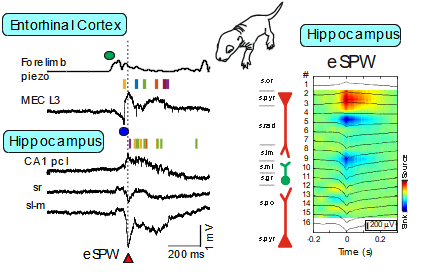
Early research into the activity patterns of developing neuronal networks relied on ex vivo preparations from neonatal rodents. These studies revealed spontaneous network bursts—such as hippocampal Giant Depolarizing Potentials (GDPs) and early network oscillations—and helped identify their underlying mechanisms (Ben-Ari et al., 2007; Khalilov et al., 2015; Leinekugel et al., 1997). However, the physiological relevance of these patterns to neonatal brain activity in vivo remained unclear. To bridge this gap, we developed a platform combining multisite intracortical silicon probe recordings and patch-clamp techniques in neonatal rats. This allowed us to characterize activity patterns expressed in vivo, including early sharp waves (eSPWs) in the hippocampus (Leinekugel et al., 2002). These eSPWs are triggered by spontaneous movements and involve activation of the entorhinal cortex (Valeeva et al., 2019). Using this approach, we also identified movement-driven oscillatory patterns in the primary somatosensory cortex (S1) and thalamus (Akhmetshina et al., 2016; Khazipov et al., 2004; Minlebaev et al., 2011) and in the spinal cord (Inacio et al., 2016). Similarly, we observed bursts driven by retinal waves in the visual cortex (Colonnese et al., 2010; Colonnese & Khazipov, 2010; Hanganu et al., 2006). Two-photon imaging has revealed synchronized activity even earlier in development, in the embryonic mouse cortex (Yuryev et al., 2018). Further translation of these findings to the human neonates resulted in elaboration of protocols of sensory exploration of the neonate in different sensory modalities (somatosensory, visual and auditory), that was achieved through long-standing collaboration with the group of Anna Kaminska at Necker Hospital (Paris) (Chipaux et al., 2013; Colonnese et al., 2010; Kaminska et al., 2018; Milh et al., 2007).
Networks organization in the developping primary somatosensory cortex
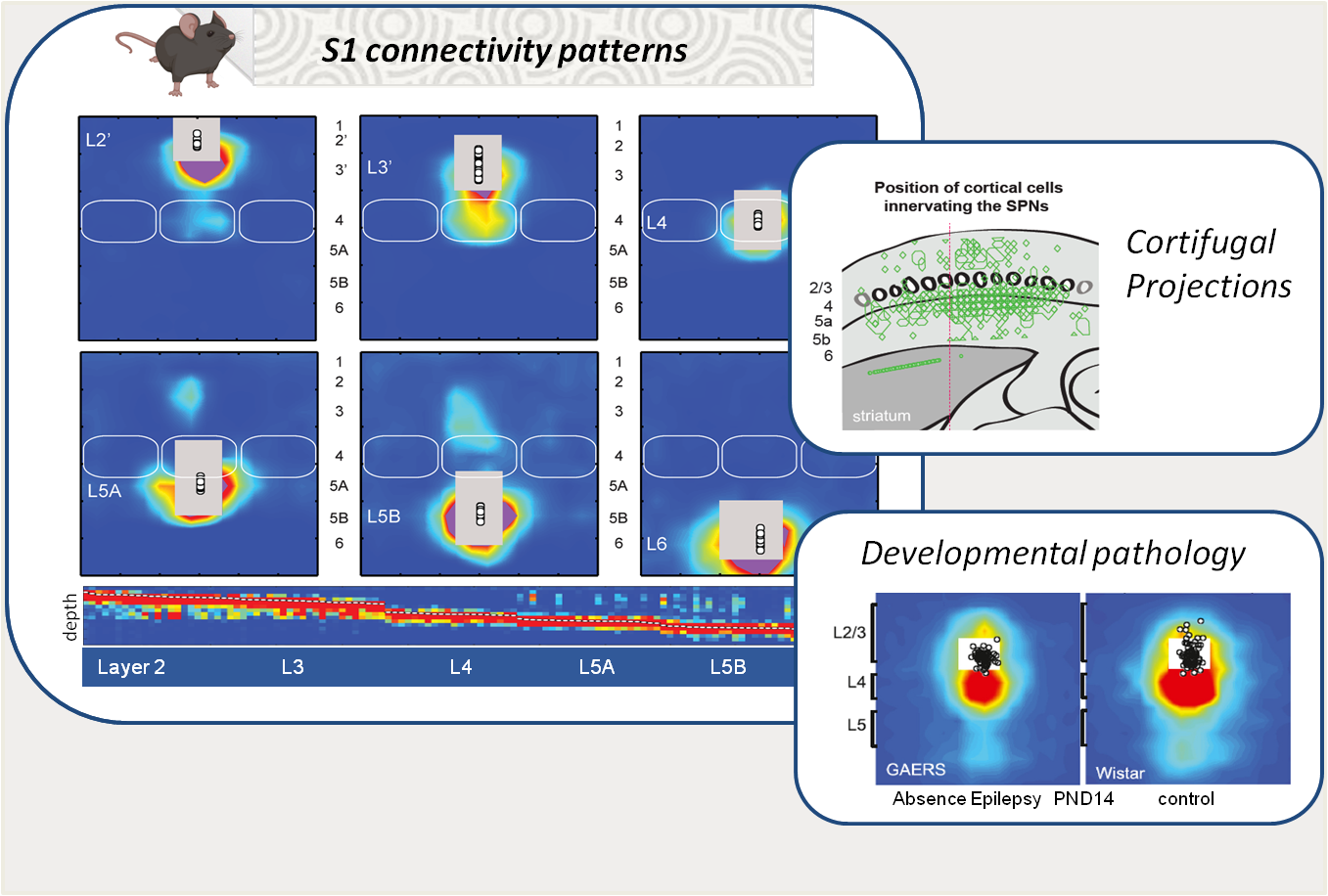
Ingrid Bureau & Juliette Contadini
The primary somatosensory cortex (S1) is structured into layers and columns, which are differentially innervated by local neurons and by upstream structures involved in tactile information processing. Its architecture, composed of first- and second-order pathways, plays a fundamental role in sensory integration as well as in the generation of motor responses, particularly through its projections to motor control structures such as the striatum. Using functional mapping approaches (e.g., laser scanning photostimulation and patch-clamp), we study the development of these networks in rodents, their plasticity in relation to learning processes, and their alterations in pathological models. Recently, our work revealed transient innervation patterns that are present during normal development but absent in a model of absence epilepsy (link). In addition, we identified a specific pattern of cortical output innervation onto striatal neurons that resolves a long-standing paradox: the coexistence of diffuse corticostriatal innervation with the strong sensory selectivity observed in striatal neurons (link).
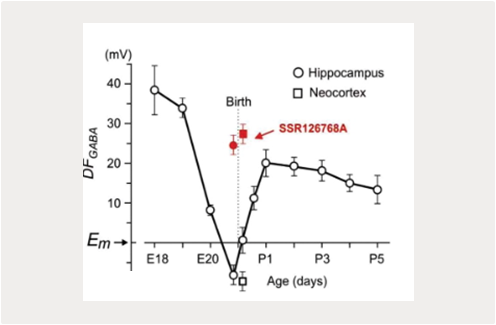
Depolarizing actions of GABA in development and disease
Rustem Khazipov & Ilgam Khalilov
Our previous studies identified instrumental roles for depolarizing and excitatory actions exerted by GABA on immature neurons, and synergistic actions of GABA and glutamate In GDPs generation in vitro (Ben-Ari et al., 2007; Khalilov et al., 2015; Leinekugel et al., 1997). Various aspects of depolarizing GABA actions on immature neurons have been characterized including the oxytocin mediated perinatal switch in GABA actions, dualism and dynamic activity-dependent changes in GABA actions at the network level (Khalilov et al., 1999; Khalilov et al., 2017; Tyzio et al., 2006; Valeeva et al., 2010). Yet, these findings were mainly obtained using in vitro preparations of brain slices or intact preparations in vitro. Explorations in the Intact brain of neonatal rodents in vivo yielded different results compatible with rather inhibitory than excitatory actions of GABA (Colonnese et al., 2010; Minlebaev et al., 2007; Valeeva et al., 2016). Interestingly, depolarizing shifts in GABA reversal potential and acquisition of the “immature” excitatory GABA phenotype have also been suggested to participate in various disease models Including epilepsy (Dubanet et al., 2021b; Khalilov et al., 2003; Kourdougli et al., 2017; Nardou et al., 2009; Pallud et al., 2014) and brain trauma (Dzhala et al., 2012; Goubert et al., 2019). These changes in GABA actions were associated with down-regulation of KCC2-mediated chloride extrusion, and could be partly restored using selective blocker of chloride-loading NKCC1 co-transporter bumetanide. Nevertheless, as regarding development, the relevance of excitatory GABA in these pathological conditions in vivo is still a matter of active debate, and was not confirmed by the only direct investigation performed in vivo, using the methodology developed by our group (Dubanet et al., 2021b).
Developmental brain plasticity
Claudio Rivera & Aurelie Carabalona
The development of the cerebral cortex is an extremely complex process that is divided into three stages: the proliferation of neuronal progenitors, neuronal migration, and the maturation of neural networks. These different steps require a significant remodeling of the membranes and a dynamic cytoskeleton. Little is known about the molecular mechanisms underlying membrane dynamics during neuronal migration and morphogenesis.
Cell membrane curvature is a micro morphological change involved in many important cellular processes including endocytosis, exocytosis, and migration. Recent studies demonstrated that the members of an extended protein family, characterized by the presence of a membrane binding and deforming BAR (Bin– Amphiphysin– Rvs) domain function at the interface between the actin cytoskeleton and plasma membrane during the formation of membrane protrusions or invaginations (Doherty and McMahon, 2008; Frost et al., 2009; Takano et al., 2008). These proteins can either generate positive membrane curvature to facilitate the formation of plasma membrane invaginations (e.g. BAR, N-BAR and F-BAR domain proteins) or induce negative membrane curvature to promote the formation of plasma membrane protrusions (I-BAR and IF-BAR domain proteins) (Guerrier et al., 2009; Mattila et al., 2007). These proteins can interact with phosphatidylinositol-4,5-biphosphate (PI(4,5)P2)-rich membranes through their BAR domain and also with actin cytoskeleton through Wasp Homology-2 domain (WH2). Regulation of the actin cytoskeleton is well described in neurons: neuronal migration, axonal and dendritic extension and guidance, dendritic spines formation (Spillane et al., 2011).Indeed, we have recently discovered that Mim, a member of the I-BAR protein family, is fundamental in the membrane bending leading to neuronal filopodia formation, and plays an important role in spine morphology (Saarikangas et al., 2015). However, regulation of the actin cytoskeleton in glial cells has remained poorly understood, and only one glia-specific regulator has been reported so far, the I-BAR protein named Abba (Saarikangas et al., 2008).
During embryonic development, Abba is highly expressed in the radial glial progenitor cells. Despite Abba’s potential role in neuronal development, very little is known about this protein, including how this protein may lead to neurological pathology (Alazami et al., 2015).
Our research proposal aims to phenotypically and mechanistically characterize a potentially new syndrome of Abba-dependent brain malformation:
I) determine the role of Abba on RGP cell fate and brain development.
II) explore Abba’s potential partners in order to define the molecular mechanism of Abba’s function in brain development.
II) correlate our results with the human pathology thanks to our ongoing collaboration with clinicians.
The mechanisms controlling the dynamic changes in the morphology of the cell are starting to be disentangled. This novel and growing field of research is slowly finding its way into developmental neurobiology and will have a profound impact on the way we understand the development of the nervous system.
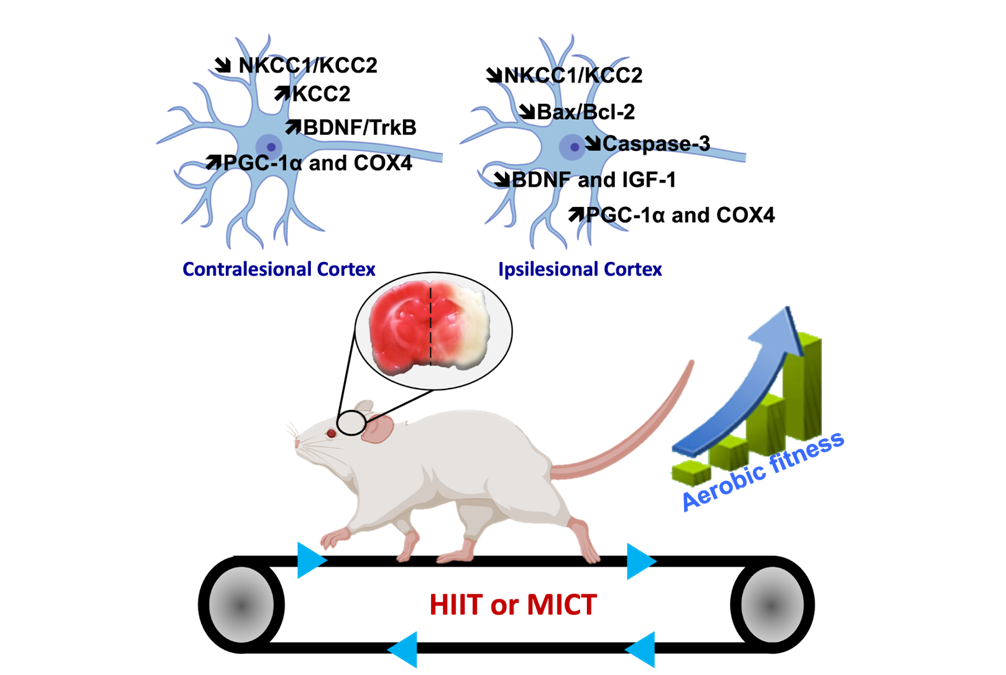
Healthy and pathological brain & exercise
JÉRÔME LAURIN, CLAUDIO RIVERA & CHRISTOPHE PELLEGRINO
The present research topic focuses on the impact of different endurance exercises on cortical and hippocampal functions, as well as on cognitive and sensorimotor functions, in healthy subjects (both young and elderly) and those who have suffered a stroke. The proposed approach to this subject involves the utilisation of multidisciplinary measures across diverse study models, encompassing behavioural (sensorimotor, cognitive, depressive state), molecular (to delineate the molecular signatures of each training method), and electrophysiological (to facilitate a more functional approach) domains.
It is the objective of this study to define the role of the major molecular players in brain plasticity, with a particular focus on neurotrophic factors, their interaction with chloride ion co-transporters, and neurogenesis. This objective is based on the following different measures. Lactate and myokines represent two of the factors involved in brain plasticity, which are being used in order to identify the mechanisms specific to each training method, thus justifying their specific benefits.
Post traumatic depression and reactive plasticity
Christophe Pellegrino & Marine Tessier
Traumatic brain injuries (TBI) are the leading injury-related cause of death and permanent disability. In France, the cerebrovascular accident (CVA) and traumatic insult are the 3rd cause of the mortality after cancers and cardio-vascular diseases. 20% of patients suffering from brain insults die within one month after injury. 75 % of CVA or TBI survivors suffer from the accident their whole life more dramatically, up to 25% will never return to professional life.
Neurodegeneration, as observed in traumatic brain injury (TBI), is a devastating sequel common to many neuropathological conditions. TBI-induced neurodegeneration is also a major risk factor for epilepsy and major depressive disorders but the mechanisms of post-traumatic depression are poorly understood. A current view is that the brain reacts to pathological insults by activating developmental-like programs for survival, regeneration and replacement of damaged neurons. For instance, we have shown that only after injury mature central neurons become dependent on brain-derived neurotrophic factor (BDNF) trophic support for survival. Interestingly, secondary neurogenesis happening in the hippocampus is a key player in the settling-up of post-traumatic depression. Our aims are:
I) Study the mechanism of secondary neurogenesis after trauma;
II) Analyze the neuronal networks and more precisely the GABA neurotransmission in pathological condition;
III) Study the inflammatory processes at the basis of neuronal death;
IV) Study the functional consequences of such insults from gene to behavior.
However the intrinsic mechanisms causing trauma-induced changes in GABAA transmission and consequent rearrangement of post-traumatic neuronal networks are not known. We are interested in understanding how neuro-inflammtion processes could participate in the establishment of long-term consequences of brain trauma through changes in secondary neurogenesis and synaptic integration.
It is important to realize that the delayed progressive reorganization may stand for the delayed sequelae commonly observed in patients suffering from different forms of brain injury e.g. post-traumatic depression.
Our team is well known for its achievements in the rapidly expanding research on neuronal ion regulation and its role in signaling, development, plasticity and pathophysiology (Pallud et al., 2014; Pellegrino et al., 2011; Payne et al., 2003; Blaesse et al., 2009; Rivera et al., 2005). Combining molecular biology and electrophysiology, we discovered that the neuronal K-Cl cotransporter, KCC2, plays a key role in the developmental shift of GABAA-mediated neurotransmission from depolarizing to hyperpolarizing (Rivera et al., 1999), and showed that activity-dependent regulation of KCC2 expression requires BDNF signaling (Rivera et al., 2002). The ongoing work has revealed a central role of CCCs in neuronal plasticity and disease mechanisms, particularly epilepsy and trauma (Goubert et al., 2019; Pallud et al., 2014; Huberfeld et al., 2007). Work on the molecular mechanisms underlying these effects has led to the identification of an increasing number of novel, fundamental interactions among growth factors and CCCs (de Koninck Y et al., 2007; Viemari et al., 2011).
Experimental approaches
We use a combination of electrophysiology and imaging (intrinsic optical signaling, surface-sensitive imaging) to study brain activity in vivo as well as on acute slices in vitro but also approaches at the cellular and molecular level.
Keywords
Neonate, Electroencephalogram, Cortex, Barrel system, Visual, GABA, Seizure, Hypoxia, Pain, Oxytocin, brain plasticity, cognition
Collaborators
Prof. G. Buzsaki (Rutgers, USA) on the early brain activity and sensory processing in the behaving neonatal rats;
Prof. G.L. Holmes (Harvard-Dartmouth, USA) on the paroxysmal activity in the developing brain and the consequences of the neonatal seizures;
Dr. Anna Kaminska and C.Chiron (Saint Vincent Paul, Paris) on the ontogenesis of human EEG;
Prof. O.Delalande (Rotschild Fondation, Paris) on the neuronal and network properties of immature human epileptic cortex;
M. Mazzuca (Necker) and Prof. R.Giniatullin (Kuopio University, Finland) on the analgesic actions of oxytocin at birth;
Drs. R.Nabout and C.Chiron (Necker, Paris) on the animal model of Dravet syndrome;
Dr. G Huberfeld (Necker) Epilepsy
Dr. A. Sirota (Tubingen University, Germany) on the signal processing;
The team is also involved in a collaboarative project with the Laboratory of the Kazan Federal University, Russia which is supported by the Grant of Government of the Russian Federation.
.
Past members
Inacio Ana-Rita (post-doc)
Carreno Maria Isabel (PhD student)
Dubanet Olivier, PhD (PhD student)
Medrano Maria Carmen (post-doc)
Bony Guillaume (post-doc)
Leinekugel Thomas (Ingénieur)
Aurelie Carabalona (post-doc)
Coralie Discala (post-doc)
Laura Caccialupi (post-doc)
Amina Rezzag (PhD student)
Florence Molinari, CRCN INSERM
Marine Tessier (PhD student)
Amandine Consumi (PhD student)
Azat Gainutdinov (PhD student)
Milh M (PhD student)
Tyzio R. (postdoc)
Kourdougli N (PhD student)
Goubert E (PhD student)
Hanganu I (post doc)
Colonnese M (post doc)
Melyan Z (post doc)
Morozova E (student)
Lamsa K (post doc)
Congar P. (PhD student)
Dzhala V. (postdoc)
Petanjek Z (postdoc)
Kenza Amroune (PhD student)
Internships
Our team has open positions for undergraduate students, PhD students and postdoctoral fellows. Applications should be sent to:
roustem.khazipov@inserm.fr
claudio.rivera@inserm.fr
christophe.pellegrino@inserm.fr
ingrid.bureau@inserm.fr















